Our research has been financially supported by NIH, USDA, and SLU funding programs.
Protein domain-centric computational strategies: The past decade has seen an unprecedented growth of genomic data from a wide range of organisms including bacterial, archaea and eukaryotes. This offers us a great opportunity to ask and answer diverse biological questions which were previously impossible. Our research focus is to mine this massive genomic data for novel biological discoveries through a unique domain-centric bioinformatics strategy. Protein domains are the conserved modules of proteins which retain functional and structural identity over large evolutionary time spans. Here we use the protein domain as a key concept for a series of large-scale computational analyses such as sequence and structural analysis, comparative genomics, and phylogenetic inferences.
- At the protein sequence and structural level, we study their structural and functional determinants such as conserved motifs, structural features, conserved folds and domain architectures to make inferences regarding their biochemical or biological functions.
- At the genome level, we study genome organization or gene neighborhood (including bacteria operonic structure) to identify biologically related interactions between domains and reconstruct novel biochemical pathways or systems.
- Finally, at the organismal level, we establish protein domain organizations and dynamics across life and try to delineate evolutionary principles shaping the speciation and major genetic and biochemical transition of life.
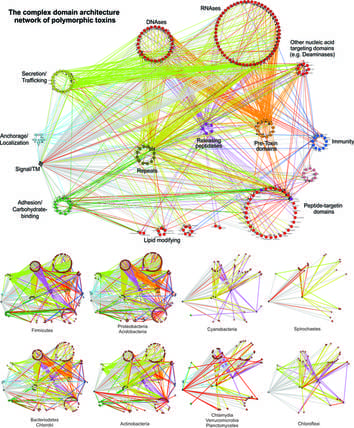
Discovery of novel toxin systems in bacteria: Protein toxins are the main players mediating bacterial interactions with other bacterial strains/species, bacteriophages and their hosts, including humans. Most known toxins, for example those from toxin-antitoxin and CRISPR systems, are involved in interactions between bacteria and phages or plasmids. Our primary contribution is the discovery of novel Polymorphic Toxin Systems deployed by all major bacterial lineages in facilitating interactions between bacterial strains/species or interactions between bacteria and their various hosts, both prokaryotic and eukaryotic.
- We have defined two underlying principles of these toxin systems in genomic organizations and toxin domain architectures.
- We uncovered over 150 novel toxin domains, 90 novel immunity families and many other components involved in secretion and toxin releasing processes, which together annotated over 40,000 previously-unknown bacterial toxin components.
- We provided functional predictions for many toxin domains, including divergent versions of peptidases, nucleases, deaminases, ADP-ribosyltransferases, and other lipid and carbohydrate-modifying enzymes.
- These discoveries have served as the underlying framework for many experimentalists to study the mechanism of bacterial conflicts.
- Related publications: NAR 2011a; NAR 2011b; Biol Direct 2012; Curr Opin Struct Biol 2014; Curr Top Microbiol Immunol 2015; mBio 2019; Microbiol Spectrum 2021; CSBJ 2022; Protein Sci 2023; Nature, 2024.
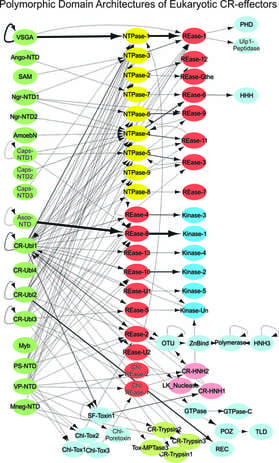
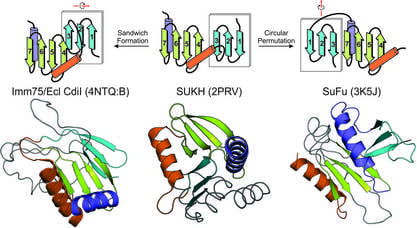
Novel effector systems underlying eukaryotic species interactions: Like bacteria, many eukaryotic species display very complex relationships and interactions with other species in conflict settings including pathogenesis, predation, parasitism, and symbiosis. However, their molecular foundations are largely unknown. In our recent research, we identified a new class of Polymorphic Effector Systems present in many eukaryotic species, analogous to polymorphic toxin systems described above in bacteria.
- The effector proteins includes the previously well-studied yet functionally poorly-understood Crinkler effectors of oomycetes and pathogenic fungi, RHS proteins of parasitic trypanosomatids, the MEDEA proteins of Tribolium which mediate post-zygotic killing, and proteins in the rhizarian Plasmodiophora and symbionts like Capsaspora, and members from free living eukaryotes such as several plant species.
- The majority of these proteins have a characteristic domain architecture with an N-terminal “Header” domain, which is involved in traffic of the protein, followed by one or more diverse C-terminal domain(s). The C-terminal domains belong to many novel versions of nucleases, kinases,and peptidases, and are predicted to be the toxicity determinants.
- These systems are the first widespread type of molecular weaponry reported to exist across diverse eukaryotic conflicts. They also provide a firm molecular basis to understand diseases such as those caused by Crinkler effectors, and also uncover potential novel pathogenic determinants in human diseases caused by kinetoplastids.
- Related publication: NAR2016
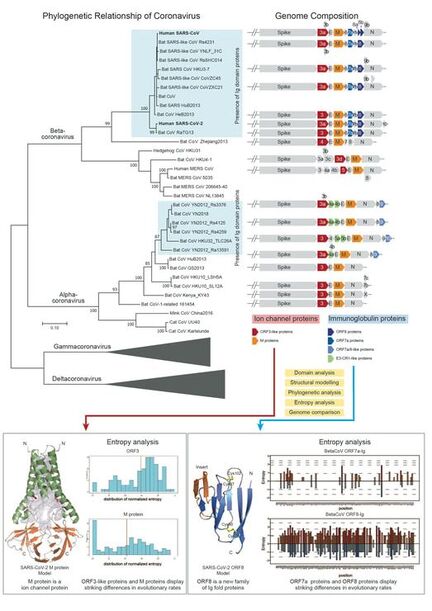
Function and evolution of SARS-CoV-2 and related viruses: The ongoing COVID-19 pandemic strongly emphasizes the need for a better understanding of the function and evolution of its causative agent SARS-CoV-2. Despite intense scrutiny, several proteins of SARS-CoV-2 remain enigmatic. By using a series of dedicated computational methods, we have successfully uncovered several previously unrecognized families of immunoglobulin (Ig) proteins and ion channel proteins in SARS-CoV-2 and many other viruses.
- The novel Ig proteins include the mysterious ORF8 proteins from SARS-CoV/SARS-CoV-2 related viruses, many proteins from alpha-CoVs and unrelated animal viruses. We show that the ORF8 proteins from the SARS-CoV/SARS-CoV-2 clade are rapidly evolving, which suggests that they might function as immune modulators to delay/attenuate the host immune response against viruses.
- We unified the SARS-CoV ORF3a family with several families of viral proteins, including ORF5 from MERS-CoVs, ORF3c from beta-CoVs, ORF3b in alpha-CoVs, most importantly, the Matrix proteins from all CoVs, and more distant homologs from other nidoviruses. We presented computational evidence that these viral families utilize specific conserved polar residues to constitute an aqueous pore within the membrane-spanning region. This suggest that the novel coronavirus Matrix/ORF3 ion channel proteins might play a role in virion assembly and membrane budding.
- Related publications: mBio 2020; Virus Evolution 2021
Understanding molecular foundations of human diseases: Recent Genome-Wide Association Studies (GWAS) have identified many mutants of genes involved in heritable human diseases; however, the functions of proteins encoded by many of these genes or their mutants are largely unknown. We utilized computational methods to characterize many disease-related proteins of unknown biochemical function. We have contributed to research on several human diseases, including ciliopathies, neurodegenerative diseases (ALS/FTD, CMT), heart disease and cancer.
- Human ciliopathies, including Nephronophthisis (NPHP), Meckel-Gruber syndrome (MKS), and Bardet-Biedl syndrome (BBS), are characterized by a defective ciliary structure. We discovered that many key ciliopathy-related proteins contain distinct transglutaminase-like peptidase domains and many novel C2 domains bearing membrane localization activities. These revealed that ciliopathies, although arising from mutation of different genes, share a common mechanistic basis related to disruption of membrane interactions. By working with Dr. Rajat Rohatgi, we show that the disruption of the interaction of a ciliary protein complex--EFCAB7, IQCE, Evc and Evc2— is the molecular foundation of Ellis van Creveld and Weyers syndromes. Related publications: Gene 2010; Cell Cycle 2012; Dev Cell 2014
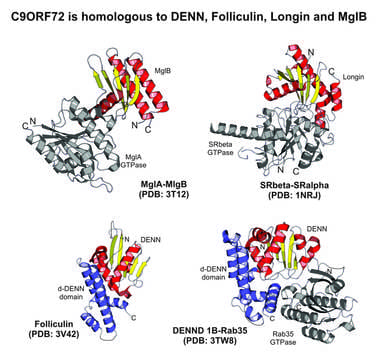
- In our research on neurodegenerative diseases, we revealed that the mysterious protein, C9ORF72, implicated in amyotrophic lateral sclerosis (ALS) and fronto-temporal dementia (FTD), defines a novel DENN-like GDP-GTP exchange factor in a specific Rab-dependent vesicular trafficking process. This suggest that ALS might be caused by a defect of vesicular transport in neurons. Related publications: Front Genet 2012; Neurol Genet 2016
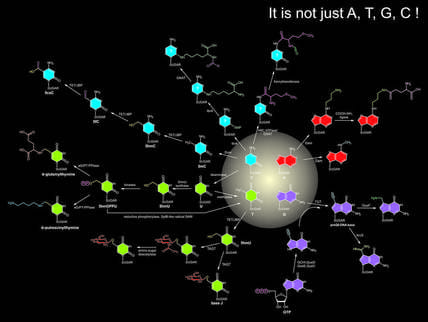
Uncovering codes of DNA modifications: It has been well known that bases in nucleic acids can be further catalytically modified, which represent a new layer of information beyond the coding capacity of conventional bases. DNA-modifications, such as cytosine and adenine methylation, have a critical role as epigenetic marks that direct DNA repair, chromatin organization, gene silencing, and repression of selfish DNA elements. While a large number of modified bases have been identified, many of the enzymes generating them still remain undiscovered. By working with Aravind and Laks at NCBI, we aim to reconstruct biochemical systems that catalyze modifications of nucleic acids in different organisms.
- We established the origins of major eukaryotic DNA-modifying enzymes from bacteriophage DNA modification systems.
- We reconstructed several key pathways which catalyze yet unknown modifications of DNAs, including the long-mysterious glycosyltransferase generating base-J in kinetoplastids.
- We discovered lineage-specific expansions of TET/JBP genes involved in oxidization of 5-methylcytosine in major clades of basidiomycete fungi, and established that the expansion was mediated by a novel epigenetically-controlled transposon mechanism.
- Related publications: NAR2013; PNAS 2014; CSHPB 2014; BioEssays 2016; PNAS2019
